3 頭孢替唑鈉藥典標準
3.1 品名
3.1.1 中文名
3.1.2 漢語拼音
Toubaotizuona
3.1.3 英文名
Ceftezole Sodium
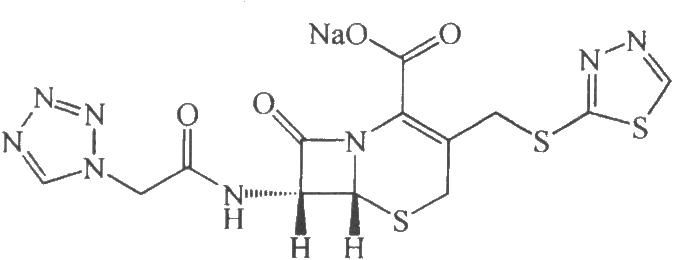
3.2 結構式
3.3 分子式與分子量
C13H11N8O4NaS3 462.47
3.4 來源(名稱)、含量(效價)
本品爲(6R,7R)-3-[[(1,3,4-噻二唑-2-基)硫]甲基]-7-[(1H-四唑-1-基)乙酰氨基]-8-氧代-5-硫雜-1-氮雜雙環[4.2.0]辛-2-烯-2-甲酸鈉鹽。按無水物計算,含頭孢替唑(C13H12N8O4S3)不得少於90.0%。
3.5 性狀
本品爲白色至淡黃色粉末或結晶性粉末;無臭,有引溼性。
3.5.1 比旋度
取本品,精密稱定,加水溶解並定量稀釋製成每1ml中約含0.1g的溶液,依法測定(2010年版藥典二部附錄Ⅵ E),比旋度爲-5°至-9°。
3.5.2 吸收係數
取本品,精密稱定,加水溶解並定量稀釋製成每1ml中約含16μg的溶液,照紫外-可見分光光度法(2010年版藥典二部附錄Ⅳ A),在272nm的波長處測定吸光度,吸收係數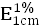 爲270~300。
爲270~300。
3.6 鑑別
(1)取本品,加水製成每1ml中約含16μg的溶液,照紫外-可見分光光度法(2010年版藥典二部附錄Ⅳ A)測定,在272nm的波長處有最大吸收。
(2)在含量測定項下記錄的色譜圖中,供試品溶液主峯的保留時間應與對照品溶液主峯的保留時間一致。
(3)本品的紅外光吸收圖譜應與對照的圖譜(《藥品紅外光譜集》1126圖)一致。
(4)本品顯鈉鹽鑑別(1)的反應(2010年版藥典二部附錄Ⅲ)。
3.7 檢查
3.7.1 酸度
取本品,加水製成每1ml中含0.1g的溶液,依法測定(2010年版藥典二部附錄Ⅵ H),pH值應爲4.5~6.5。
3.7.2 溶液的澄清度與顏色
取本品5份,各0.6g,分別加水5ml溶解後,溶液應澄清無色;如顯渾濁,與1號濁度標準液(2010年版藥典二部附錄Ⅸ B)比較,均不得更濃;如顯色,與黃色或黃綠色6號標準比色液(2010年版藥典二部附錄Ⅸ A第一法)比較,均不得更深。
3.7.3 有關物質
取本品適量,加水溶解並稀釋製成每1ml中約含1mg的溶液,作爲供試品溶液;精密量取適量,用水定量稀釋製成每1ml中約含10μg的溶液,作爲對照溶液。照含量測定項下的色譜條件,取對照溶液20μl注入液相色譜儀,調節檢測靈敏度,使主成分色譜峯的峯高約爲滿量程的25%,再精密量取供試晶溶液與對照溶液各20μl,分別注入液相色譜儀,記錄色譜圖至主成分峯保留時間的2.5倍。供試品溶液色譜圖中如有雜質峯,單個雜質峯面積不得大於對照溶液主峯面積(1.0%),各雜質峯面積的和不得大於對照溶液主峯面積的1.5倍(1.5%)。
3.7.4 頭孢替唑聚合物
照分子排阻色譜法(2010年版藥典二部附錄Ⅴ H)測定。
3.7.4.1 色譜條件與系統適用性試驗
用葡聚糖凝膠G-10(40~120μm)爲填充劑,玻璃柱內徑1.0~1.4cm,柱長30~40cm,流動相A爲pH 7.0的0.075mol/L磷酸鹽緩衝液[0.075mol/L磷酸氫二鈉溶液-0.075mol/L磷酸二氫鈉溶液(61:39)];流動相B爲水,流速爲每分鐘1.5ml,檢測波長爲254nm。取0.4mg/ml藍色葡聚糖2000溶液100~200μl注入液相色譜儀,分別以流動相A、B爲流動相,記錄色譜圖。理論板數按藍色葡聚糖2000峯計算均不低於300,拖尾因子均應小於2.0,在兩種流動相系統中藍色葡聚糖2000峯的保留時間的比值應在0.93~1.07之間,對照溶液主峯與供試品溶液中聚合物峯與相應色譜系統中藍色葡聚糖2000峯的保留時間的比值均應在0.93~1.07之間,稱取本品約0.2g,置10ml量瓶中,用0.4mg/ml藍色葡聚糖2000溶液溶解並稀釋至刻度,搖勻,量取100~200μl注入液相色譜儀,用流動相A進行測定,高聚體的峯高與單體與高聚體之間的谷高比應大於2.0,另以流動相B爲流動相,精密量取對照溶液100~200μl,連續進樣5次,峯面積的相對標準偏差應不大於5.0%。
3.7.4.2 對照溶液的製備
取頭孢替唑對照品適量,精密稱定,加水溶解並定量稀釋製成每1ml中約含頭孢替唑25μg的溶液。
3.7.4.3 測定法
取本品約0.3g,精密稱定,置10ml量瓶中,加水溶解並稀釋至刻度,搖勻。立即精密量取100~200μl注入液相色譜儀,以流動相A爲流動相進行測定,記錄色譜圖。另精密量取對照溶液100~200μl注入液相色譜儀,以流動相B爲流動相進行測定,記錄色譜圖。按外標法以峯面積計算,含頭孢替唑聚合物以頭孢替唑計,不得過0.05%。
3.7.5 水分
取本品,照水分測定法(2010年版藥典二部附錄Ⅷ M第一法 A)測定,含水分不得過5.0%。
3.7.6 可見異物
取本品5份,每份各4.0g,用微粒檢查用水溶解,依法檢查(2010年版藥典二部附錄Ⅸ H),應符合規定。
3.7.7 不溶性微粒
取本品3份,用微粒檢查用水製成每1ml中含頭孢替唑60mg的溶液,依法檢查(2010年版藥典二部附錄Ⅸ C),每1g樣品中含10μm以上的微粒不得過6000粒,含25μm以上的微粒不得過600粒。
3.7.8 細菌內毒素
取本品,依法檢查(2010年版藥典二部附錄Ⅺ E),每1mg頭孢替唑中含內毒素的量應小於0.075EU。
3.7.9 無菌
取本品,加無菌水適量溶解後,全部轉移至不少於500ml的0.9%無菌氯化鈉溶液中,用薄膜過濾法處理後,依法檢查(2010年版藥典二部附錄Ⅺ H),應符合規定。
3.8 含量測定
照高效液相色譜法(2010年版藥典二部附錄Ⅴ D)測定。
3.8.1 色譜條件與系統適用性試驗
用十八烷基硅烷鍵合硅膠爲填充劑;以枸櫞酸溶液(取枸櫞酸3g,加水溶解並稀釋至900ml)-乙腈(90:10)爲流動相;檢測波長爲254nm。取頭孢替唑對照品約25mg,置25ml量瓶中,加0.1mol/L氫氧化鈉溶液1ml使溶解,放置1分鐘,加水10ml,再加0.1mol/L鹽酸溶液1ml中和,用水稀釋至刻度,搖勻,得每1ml中約含頭孢替唑1mg與其降解雜質的混合溶液(其中相對保留時間0.8與1.7處雜質的量約爲0.5%和2%),取20μl注入液相色譜儀,記錄色譜圖,頭孢替唑峯保留時間約爲13分鐘,頭孢替唑峯與其相對保留時間0.8處雜質峯的分離度應不小於4.0。
3.8.2 測定法
取本品適量,精密稱定,加水溶解並定量稀釋製成每1ml中約含0.2mg的溶液,精密量取20μl注入液相色譜儀,記錄色譜圖;另取頭孢替唑對照品適量,同法測定。按外標法以峯面積計算供試品中C13H12N8O4S3的含量。
3.9 類別
β-內酰胺類抗生素,頭孢菌素類。
3.10 貯藏
3.11 製劑
3.12 版本
《中華人民共和國藥典》2010年版