1 拼音
huà xué yào wù yuán liào yào zhì bèi hé jié gòu què zhèng yán jiū de jì shù zhǐ dǎo yuán zé
《化學藥物原料藥製備和結構確證研究的技術指導原則》由國家食品藥品監督管理局於2005年3月18日國食藥監注[2005]106號發佈。
指導原則編號:【H】GPH2-1
2 一、化學藥物原料藥製備研究的技術指導原則
2.1 (一)概述
原料藥的製備是藥物研究和開發的基礎,是藥物研發的起始階段,其主要目的是爲藥物研發過程中藥理毒理、製劑、臨牀等研究提供合格的原料藥,爲質量研究提供信息,通過對工藝全過程的控制保證生產工藝的穩定、可行,爲上市藥品的生產提供符合要求的原料藥。
本指導原則旨在反映原料藥製備研究的基本規律,並遵循該規律進行原料藥的研發,確定符合相關法規的、科學的基本技術要求,爲藥物研發者在原料藥製備研究中提供基本的技術指導,同時使藥物研發者對在藥品評價過程中所需要關注的問題有一個清楚的認識。
本指導原則主要內容爲概述、研究的一般過程、研究的基本內容、名詞解釋、參考文獻等。概述就本指導原則起草的目的意義、適用範圍等方面進行介紹。研究的一般過程爲藥物研發者提供原料藥製備研究的通用規律,使藥物研發者對藥物研發過程有一個整體的認識。研究的基本內容主要對工藝的選擇、起始原料和試劑的要求、工藝數據的積累、中間體的要求、工藝的優化與中試、雜質的分析、“三廢”的處理和工藝的綜合分析等方面進行了闡述,其研究內容基本上是按照原料藥製備的研發過程進行設置的,從實驗室階段到工業生產階段均提出了相應的要求,強調了對工藝過程控制的重要性,目的是使藥物研發者按照以上要求進行研究可以得到一個合理、可行的生產工藝;名詞解釋對本指導原則中涉及的專有名詞進行解釋,以免引起歧義;參考文獻給出本指導原則所採用的文獻依據。
本指導原則是一個通用的原則,適用於經化學全合成或半合成以及從動、植物中提取的原料藥的研製,包括新藥、進口藥和已有國家標準的藥物。經微生物發酵得到的藥物也可參考本指導原則的相關要求。
需要說明的是,在藥物研發過程中,由於藥物自身的特性,存在很多特殊情況,並且隨着學科的發展,新技術和新方法不斷出現,會遇到很多目前難以預料的問題,因此本指導原則只是給予藥物研發者在原料藥製備研究中應關注的基本問題,藥物研發者亦可根據原料藥研發的實際情況,採用其他更有效的方法和手段,但是必須符合藥物研發的規律,並提供科學合理的依據。
2.2 (二)原料藥製備研究的一般過程
原料藥製備研究是一個複雜的過程,存在很多特殊的情況,但均應遵循一般規律性的要求,即工藝可行、穩定,能夠工業化生產,同時能製備出質量合格的原料藥,因此原料藥製備的研究必須要遵循共同的原則。本部分的目的在於闡明原料藥研發過程中共同的基本規律,爲藥物研發者提供原料藥製備研究的通用規則,使研發者不僅對原料藥製備研究的全過程有一個整體的理解,而且對其中每一階段的目的有清晰的認識,以便在藥物研發中做到有的放矢、科學穩妥、高效快捷地開展研究並獲得符合要求的原料藥。
原料藥製備研發過程一般包括以下六個階段:
1、確定目標化合物:通過文獻調研、藥效學篩選實驗或其它有關基礎研究工作,確定擬研發的目標化合物。
2、設計合成路線:根據目標化合物的結構特性,參考國內外相關文獻,綜合分析,確定工藝可行、成本合理、收率相對較高的合成路線。
3、製備目標化合物:通過化學反應、生物發酵或其他方法製備出質量符合要求的目標化合物,爲產品進行結構確證、質量控制等藥學方面的研究以及藥理毒理和臨牀研究提供合格的樣品。
4、結構確證:使用物理和化學方法,確證目標化合物的結構(參見《原料藥結構確證研究的技術指導原則》)。
5、工藝優化:綜合考慮原材料獲得的難易程度、工藝路線的反應條件、環保和安全、產品的純化等對生產工藝進行優化。
6、中試研究和工業化生產:通過對中試和工業化生產工藝的研究,確定穩定、可行的工藝,爲藥物進一步研發提供符合要求的原料藥。
2.3 (三)原料藥製備研究的基本內容
1、工藝的選擇
藥物製備工藝選擇的目的是通過對擬研發的目標化合物進行文獻調研,瞭解和認識該化合物的國內外研究情況和知識產權狀況,設計或選擇合理的製備路線。對所採用的工藝進行初步的評估,也爲藥物的技術評價提供依據。
對於新的合成化學實體,根據其結構特徵,綜合考慮起始原料獲得的難易程度、合成步驟的長短、收率的高低以及反應條件、反應的後處理、環保要求等因素,確定合理的合成路線;或者根據國內外對類似結構化合物的文獻報道進行綜合分析,確定適宜的合成方法。
對於通過微生物發酵或從動、植物中提取獲得的原料藥,經對原材料和工藝過程的可控性分析,綜合考慮成本、環保要求等,確定一條產品質量可控、收率較高的工藝路線。
對於結構已知的藥物,通過文獻調研,對有關該藥物製備的研究情況進行全面的瞭解;對所選擇的路線從收率、成本、“三廢”處理、起始原料是否易得、是否適合工業化生產等方面進行綜合分析比較,選擇合理的合成路線。若爲創新路線,應與文獻報道路線進行比較。
2、起始原料和試劑的要求
在原料藥製備工藝研究的過程中,起始原料和試劑的質量是原料藥製備研究工作的基礎,直接關係到終產品的質量和工藝的穩定,可爲質量研究提供有關的雜質信息,也涉及到工業生產中的勞動保護和安全生產問題。因此,應對起始原料和試劑提出一定的要求。
2.1 起始原料的選擇原則:起始原料應質量穩定、可控,應有來源、標準和供貨商的檢驗報告,必要時應根據製備工藝的要求建立內控標準。對由起始原料引入的雜質、異構體,必要時應進行相關的研究並提供質量控制方法;對具有手性的起始原料,應制訂作爲雜質的對映異構體或非對映異構體的限度,同時應對該起始原料在製備過程中可能引入的雜質有一定的瞭解。
2.2 試劑和溶劑的選擇:一般應選擇毒性較低的試劑,避免使用一類溶劑,控制使用二類溶劑,同時應對所用試劑、溶劑的毒性進行說明,以利於在生產過程中對其進行控制,有利於勞動保護。有機溶劑選擇的詳細內容參見《化學藥物有機溶劑殘留量研究的技術指導原則》。
2.3 內控標準:由於製備原料藥所用的起始原料、試劑可能存在着某些雜質,若在反應過程中無法將其去除或者參與了反應,對終產品的質量有一定的影響,因此需要對其進行控制,制定相應的內控標準。一般要求5 對產品質量有一定影響的起始原料、試劑應制訂內控標準,同時還應注意在工藝優化和中試過程中起始原料和重要試劑規格的改變對產品質量的影響。
一般內控標準應重點考慮以下幾個方面:(1)對名稱、化學結構、理化性質要有清楚的描述;(2)要有具體的來源,包括生產廠家和簡單的製備工藝;(3)提供證明其含量的數據,對所含雜質情況(包含有毒溶劑)進行定量或定性的描述;(4)如果需要採用起始原料或試劑進行特殊反應,對其質量應有特別的要求,如對於必須在乾燥條件下進行的反應,需要對起始原料或試劑中的水分含量進行嚴格的要求和控制;若起始原料爲手性化合物,需要對對映異構體或非對映異構體的限度有一定的要求;(5)對於不符合內控標準的起始原料或試劑,應對其精製方法進行研究,以利於對工藝和終產品的質量進行控制。
通常,在工藝穩定的條件下,所採用的起始原料、試劑的質量也應相對穩定。
3、工藝數據的積累
在藥物研發過程中,原料藥的製備工藝研究是一個不斷探索和完善的動態過程,藥物研發者需要對製備工藝反覆進行試驗和優化,以獲得可行、穩定、收率較高、成本合理並適合工業化生產的工藝。在這個重複完善的過程中,積累充足的實驗數據對判斷工藝的可行性具有重要意義,同時也爲質量研究提供有關信息。因此,在藥物研發過程中,研發者應積極主動收集有關的工藝研究數據,儘可能提供充分的原料藥製備數據的報告,並對此進行科學的分析,作出合理的結論。充分的數據報告也將有利於藥品評價者對原料藥製備工藝的評價。需要說明的是,數據的積累貫穿於藥物研發的整個過程。
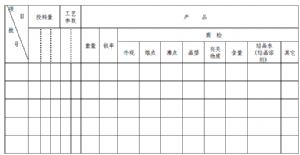
工藝數據報告應包括對工藝有重要影響的參數、投料量、產品收率及質量檢驗結果(包括外觀、熔點、沸點、比旋度、晶型、結晶水、有關物質、異構體、含量等),並說明樣品的批號、生產日期、製備地點。工藝數據報告一般分爲臨牀研究和生產兩個階段,可採用表格的形式進行彙總,參考式樣見附件。
4、中間體的研究及質量控制
在原料藥製備研究的過程中,中間體的研究和質量控制是不可缺少的部分,對穩定原料藥製備工藝具有重要意義,爲原料藥的質量研究提供重要信息,也可以爲結構確證研究提供重要依據(參見《原料藥結構確證研究的技術指導原則》)。
一般來說,由於關鍵中間體對終產品的質量和安全性有一定的影響,因此對其質量進行控制十分重要。對於新結構中間體,由於沒有文獻報道,其結構研究對於認知該化合物的特性、判斷工藝的可行性和對終產品的結構確證具有重要作用。對於一般中間體的要求可相對簡單,對其質量可以進行定量控制。有時,因終產品結構確證研究的需要,有必要對已知結構中間體的結構進行研究。
需要說明的是,中間體的質量控制應按照產品工藝路線的特點和終產品質控的需要合理選取質控項目。
4.1 新結構的中間體
一般情況下應對其結構進行確證,並對理化常數、質量控制(定性、定量)進行研究。
結構研究:一般應進行紅外、紫外、核磁共振(碳譜、氫譜,必要時進行二維相關譜)和質譜等研究,以確證該中間體的結構。理化常數研究一般包括:熔點、沸點、比旋度、溶解度等。
質量研究一般包括:性狀、異構體、有關物質、含量等。
4.2 已知結構的關鍵中間體
一般情況下應對其理化常數、質量(定性、定量)進行研究,根據結構確證研究的需要,提供相應的結構研究資料。
理化常數測定一般應包括:熔點、沸點、比旋度、溶解度等,並與文獻報道的有關數據比較。質量控制一般包括:性狀、異構體、有關物質、含量等。結構研究:如果因終產品結構確證的需要,應對其結構進行確證,並應與有關的文獻資料進行比較。
4.3 已知結構的一般中間體
一般情況下應對其理化常數進行研究,並與文獻資料進行比較,同時還應對其質量進行研究,並根據結構確證研究的需要,提供相應的結構研究資料。
質量研究一般包括:採用 TLC、HPLC 、GC 等方法,對其在反應過程中進行定量或定性控制。
結構研究:如果由於終產品結構確證研究的需要,應對其結構進行確證,並應與有關的文獻資料進行比較。
4.4 中間體的再精製
對於不符合標準的中間體,應對其再精製的方法進行研究。
5、工藝的優化與中試
在原料藥的工藝研究中,工藝的優化與中試是原料藥製備從實驗室階段過渡到工業化階段不可缺少的環節,是考察該工藝能否工業化的關鍵。
原料藥製備工藝優化與中試的主要任務是:
(1)考覈實驗室提供的工藝在工藝條件、設備、原材料等方面是否有特殊的要求,是否適合工業化生產;
(3)驗證實驗室工藝是否成熟合理,主要經濟指標是否接近生產要求;
(4)進一步考覈和完善工藝條件,對每一步反應和單元操作均應取得基本穩定的數據;
(5)根據中試研究資料制訂或修訂中間體和成品的分析方法、質量標準;
(6)根據原材料、動力消耗和工時等進行初步的技術經濟指標覈算;
(7)提出“三廢”的處理方案;
(8)提出整個合成路線的工藝流程,各個單元操作的工藝規程。一般來說,中試所採用的原料、試劑的規格應與工業化生產時一致。
從動、植物中提取的有效單體和通過微生物發酵獲得原料藥的實驗室研究和中試與合成藥物相關單元操作要求基本相似。
在工藝優化和放大過程中,中試規模的工藝在藥物技術評價中具有非常重要的意義,是評價原料藥製備工藝可行性、真實性的關鍵,是質量研究的基礎。藥物研發者應特別重視原料藥的中試研究,中試規模工藝的設備、流程應與工業化生產一致。
原料藥的工藝優化是一個動態過程,隨着工藝的不斷優化,起始原料、試劑或溶劑的規格、反應條件等會發生改變,研發者應注意這些改變對產品質量(如晶型、雜質等)的影響。因此,應對重要的變化,如起始原料、試劑的種類或規格、重要的反應條件、產品的精製方法等發生改變前後對產品質量的影響,以及可能引入新的雜質情況進行說明,並對變化前後產品的質量進行比較。
6、雜質的分析
原料藥製備過程中產生的雜質是原料藥雜質的主要來源,通過對工藝過程中產生的雜質進行詳細的研究,藥物研發者可以對工藝過程中產生的雜質有全面的認識,爲終產品的質量研究提供信息。這裏所述的雜質不包括終產品的降解物。
製備過程中產生的雜質主要有:
(1)起始原料引入的雜質
(2)副產物,如異構體
(3)副反應產生的雜質
(4)殘留溶劑、試劑和中間體
(5)痕跡量的催化劑
(6)無機雜質
雜質的研究參見《化學藥物雜質研究的技術指導原則》。
7、“三廢”的處理
在原料藥製備研究的過程中,“三廢”的處理應符合國家對環境保護的要求。在工藝研究中需對可能產生的“三廢”進行考慮,儘可能避免使用有毒、嚴重污染環境的溶劑或試劑,應結合生產工藝制訂合理的“三廢”處理方案。
在原料藥製備研究的過程中,工藝的綜合分析是重要內容之一。通過綜合分析可以使藥物研發者對整個工藝的利弊有明確的認識,同時也有利於藥品的技術評價工作。
藥物研發者在以上研究的基礎上,經對實驗室工藝、中試工藝和工業化生產工藝這三個階段的深入研究,應對整個工藝有較全面的認識,從而對原料藥的製備工藝從工藝路線、反應條件、產品質量、經濟效益、環境保護和勞動保護等方面進行綜合評價。
2.4 (四)名詞解釋
起始原料:是構成原料藥分子部分結構的化合物,能穩定、批量生產且質量可控。
中間體:在原料藥製備過程中產生的、通過進一步的化學反應才能生成原料藥的化合物。
關鍵中間體:對終產品的質量或安全性有影響的物質,其中也包括對產品質量、安全性有影響的試劑或起始原料。
已知結構中間體:已有文獻報道的中間體。
新結構中間體:尚沒有文獻報道的中間體。
雜質:是在原料藥的製備過程中,由原料、試劑、溶劑或副反應引入的與終產品結構不同的任何成分。
內控標準:是根據產品質量控制的需要,生產企業或研發單位制訂的生產工藝中的某一化合物的質量控制標準。
2.5 (五)參考文獻
1、ICH :Q3a Impurities In New Drug Substances ,1996.
2、FDA :Guidance for Industry Drug Substance Chemistry,Manufacturing,and Controls Information ,2004.
3、EMEA :Note For Guidance On Chemistry of The New Active Substance, 2003.
4、鄭筱萸. 《化學藥品和治療用生物製品研究指導原則(試行)》 。中國醫藥科技出版社, 2002,第一版。
2.6 (六)附件
工藝數據報告參考格式:
生產日期:
生產地點:
包裝:
3 二、化學藥物原料藥結構確證研究的技術指導原則
3.1 (一)概述
原料藥的結構確證研究是藥物研發的基礎,其主要任務是確認所製備原料藥的結構是否正確,是保證藥學其它方面研究、藥理毒理和臨牀研究能否順利進行的決定性因素。本指導原則是根據藥品管理法規的有關要求,通過對原料藥結構確證研究過程的分析,爲藥物結構確證研究提供基本技術要求。
本指導原則主要內容爲概述、研究的一般過程、研究的基本內容、名詞解釋、參考文獻等部分。概述主要對本指導原則起草的目的、背景、適用範圍等進行介紹。研究的一般過程主要闡述了藥物結構確證研究的基本規律,使藥物研發者對此有整體的認識。研究的基本內容對結構確證方案的確定、樣品的要求、藥物的名稱、結構式、理化常數、結構確證經常使用的方法或手段及其目的和意義、不同結構類型藥物的不同要求、綜合解析等方面進行了說明。名詞解釋對本指導原則涉及的專有名詞進行了解釋。本指導原則是一個通用的原則,適用於經化學全合成或半合成、微生物發酵以及從動、植物中提取的原料藥,包括新藥、進口藥和已有國家標準的藥品。
本指導原則僅爲基本的技術要求,隨着科學技術的發展,必然會出現新方法和新手段,因此,在藥物結構確證研究中,不應機械地照搬指導原則的方法,應結合藥物的結構特徵,採用有效的手段與方法,以達到對藥物結構準確確證的目的。
3.2 (二)原料藥結構確證研究的一般過程
隨着科學的發展和藥物研究的不斷深入,藥物的來源日趨廣泛,其結構呈現多樣性,藥物的結構確證方法也不盡相同,本部分內容是爲藥物研發者提供進行結構確證研究的通用原則,以便對藥物結構確證研究的全過程有整體的認識,達到科學、有效地證明化合物結構的目的。
結構確證的一般過程:根據化合物(藥物)的結構特徵制訂科學、合理、可行的研究方案,製備符合結構確證研究要求的樣品,進行有關的研究,對研究結果進行綜合分析,確證測試品的結構。該過程主要包括化合物的名稱,樣品的製備,理化常數的研究,樣品的測試及綜合解析等。常用的分析測試方法有紫外可見吸收光譜 (簡稱:紫外光譜 )(Ultraviolet-visible spectrophotometry,UV)、紅外吸收光譜(簡稱“紅外光譜”)(Infrared spectrophotometry,IR)、核磁共振譜(Nuclear magnetic resonance,NMR)、質譜(Mass spectrum,MS)、比旋度([α ]D)、X-射線單晶衍射(簡稱:單晶 X-衍射)(X-ray single crystal diffraction,XRSD)或/和 X-射線粉末衍射(簡稱:粉末 X-衍射)(X-ray powder diffraction,XRPD)、差示掃描量熱法(Differential scanning calorimetry,DSC)、熱重(Thermo-gravimetry,TG)等。
3.3 (三)原料藥結構確證研究的基本內容
1、研究方案的制訂
藥物結構千差萬別,製備(獲得)方法也各不相同,應根據藥物的自身結構特徵和製備(獲得)方法制訂出合理、可行的結構確證方案,纔能有效地進行藥物的結構研究。
結構確證的方案應根據藥物自身的結構特點制訂,以下對不同類型藥物的測試方案作一簡要概述。
1.1 一般藥物
採用常規方法,如元素分析(必要時採用高分辨質譜)、UV、IR、NMR、MS、熱分析(差熱或熱重)、粉末 X-衍射(XRPD )等即可確證藥物的結構。對於結構比較特殊的藥物,也可採用製備衍生物的方法間接證明藥物的結構。
對於存在順反異構的藥物,在一般結構確證的基礎上,應增加順反結構的研究。
1.2 手性藥物
除進行上述各項化學結構確證和比旋度測定外,還應採用其它有效的方法進行研究。
1.2.1 單一對映體
其絕對構型(或通過衍生物的構型)確證常用的方法爲:比旋度測定、手性柱色譜(Chiral high performance liquid chromatography 和 Chiral gas chromatography ,Chiral HPLC 和 Chiral GC)、核磁共振(NMR)、單晶 X-衍射(XRSD)以及旋光色散(Optical rotatory dispersion,ORD)、圓二色譜(Circular dichroism,CD)等。其中單晶 X-衍射爲直接方法,可提供最直接的信息。也可採用間接的方法如:在說明化合物(藥物)在反應過程中構型沒有變化的情況下,根據已知的起始原料構型、化學合成方法的立體選擇性以及中間體的結構也可間接獲得終產品(藥物)的構型信息。
1.2.2 藥物分子中含有多個不對稱因素 應對其絕對構型、對映體純度(非對映體純度)進行相關的研究,並儘可能提供更多的構型確證信息。
1.2.3 立體異構混合物 需進行各立體異構體比例的確證研究。對於已有實驗證據或文獻報道立體異構體在藥效、藥代動力學或毒理等方面有明顯不同或有相互作用的藥物,更有必要測定混合物中各組分的構型和比例。
1.2.4 外消旋體或富集對映體 可通過測定旋光度或採用手性色譜(Chiral HPLC 或 Chiral GC)及核磁共振譜等方法闡明其對映體的比例。
根據結構確證的需要,可提供成鹽前後的兩套波譜和試驗數據。對於某些波譜測定有困難或不易說明藥物結構的鹽或複合物,測定藥物的酸根或鹼基的波譜,並結合其它試驗項目亦可對其結構確證提供有效的信息。 16
在進行一般要求的各項測試基礎上,考慮以適當手段反映藥物中金屬元素的種類、存在形式和含量的確證試驗。不適於或不能測試金屬鹽本身的項目,可考慮以成鹽前的酸分子或配位體的相應測試結果進行佐證。
1.5 半合成藥物
分子中母核的結構爲已知並在可提供明確證據證明原分子母核結構在半合成全過程中未發生改變的前提下,適當簡化對母核部分結構的確證工作,僅對新引入的基團結構進行確證。
1.6 多晶型藥物
在進行一般要求的各項測試基礎上,應以適當方法獲得藥物晶型數據。藥物晶型測定常用方法爲粉末 X-衍射(XRPD)、紅外吸收光譜、熔點、熱分析、光學顯微鏡等。
該類藥物一般可分爲以下幾種情況:
1.6.1 新化學實體的藥物 應進行藥物在不同結晶條件下(溶劑、溫度、結晶速度等)是否存在多種晶型的研究。
1.6.2 已有文獻報道存在多晶型的藥物 應明確藥物晶型的類型和純度。對於混晶藥物,應測試其晶型組成(種類、比例),並與文獻數據比較。對於因晶型影響藥物的溶解性、穩定性、生物利用度和活性的藥物,在無相應藥理毒理等研究證明該晶型的安全和有效性時,應確證自制品與國外上市藥品晶型的一致性。
該類藥物在進行一般分析時,熱分析研究已經提供了藥物中的結晶水17 或結晶溶劑的信息,結合乾燥失重、水分或單晶 X-衍射(XRSD)等方法的測定結果,基本上可以達到對藥物中結晶水/溶劑以及吸附水/溶劑進行定性、定量的目的。
1.8.1 合成多肽藥物 通過氨基酸分析、質譜測定、序列分析以及肽圖測繪(含有 20 個以上的氨基酸殘基藥物)等實驗可基本獲得合成多肽藥物的結構信息。藥物結構中如有半胱氨酸,應明確其狀態(氧化態或還原態),對含有多個半胱氨酸的多肽藥物,應明確二硫鍵的正確連接位點。 如各步中間體均進行了質譜測定,可根據相關中間體的結構信息,推測出進行反應的氨基酸的種類。
質譜是多肽藥物結構確證的重要手段,紫外、紅外、核磁共振、多種流動相 HPLC、比旋度測定等方法亦可對肽的結構確證提供幫助。對於多肽藥物,應對目標物的化學純度和對映體或非對映體純度進行研究。
1.8.2 多糖類藥物 通過對單糖組成、分子量、糖苷鍵連接方法和連接位置等的分析,可獲得多糖類藥物的基本結構信息。單糖的分離和鑑定可採用紙色譜、薄層色譜、高效液相色譜、色-質聯用等技術。多糖的相對分子量及分子量分佈測定可用凝膠色譜等方法。紅外光譜、核磁共振、化學反應後產物的分析等實驗,可幫助確定糖苷鍵的連接方式及糖苷鍵的位置。
1.9 多組份藥物 應明確各組份的組成比例,對其主要成分應進行結構確證,具體方法可參照本指導原則的相關要求。
1.10 其它 上述未提及的具有特殊結構,需特殊方法進行說明、確證的藥物,可根據其結構特徵,制訂能反映藥物自身結構特徵的方法進行結構研究。
2、測試樣品的要求
在結構確證的研究中,測試樣品的純度需要進行一定的控制,只有使用符合要求的測試品進行結構研究,才能獲得藥物正確的結構信息。 一般情況下,應採用原料藥製備工藝中產品的精製方法對樣品進行精製,並採用質量標準中的方法測其純度和雜質,供試樣品的純度應大於99.0%,雜質含量應小於 0.5%。
3、結構確證研究的一般內容
3.1.1 藥物元素組成 通常採用元素分析法。這種方法可獲得組成藥物的元素種類及含量,經比較測試結果與理論結果差值的大小(一般要求誤差不超過 0.3%),即可初步判定供試品與目標物的分子組成是否一致。 對於因藥物自身結構特徵而難於進行元素分析時,在保證高純度情況下可採用高分辨質譜方法獲得藥物元素組成的相關信息。
3.1.2 紫外吸收光譜(UV) 通過對藥物溶液在可見-紫外區域內在不同波長處吸收度的測定和吸收係數(尤其是摩爾吸收係數)的計算,以及對主要吸收譜帶進行歸屬(如 K 帶、R 帶、E 帶、B 帶),可獲得藥物結構中可能含有的髮色團、助色團種類以及初步的連接方式等信息,同時對藥物的鑑別亦有指導意義。
對於髮色團上存在酸性或鹼性基團的藥物,通過在酸或鹼溶液中(常用 0.1mol/L HCl 或 0.1mol/L NaOH)最大吸收波長的測試,觀察其紫移或紅移現象,可爲上述酸性或鹼性基團的存在提供進一步的支持。
3.1.3 紅外吸收光譜(IR) 通過對藥物進行紅外吸收光譜測試,可推測出藥物中可能存在的化學鍵、所含的官能團及其初步的連接方式,亦可給出藥物的幾何構型、晶型、立體構象等信息。
固態藥物紅外測試可分爲壓片法、糊法、薄膜法,液態藥物可採用液膜法測試,氣態藥物則可採用氣體池測定。
部分含多晶型藥物在研磨和壓片過程中,其晶型可能發生變化,可改用糊法測定,同時應根據藥物的結構特點對糊劑的種類進行選擇。鹽酸鹽藥物在採用 KBr 壓片時可能會發生離子交換現象,應分別對氯化鉀壓片和溴化鉀壓片法測得的結果進行比較,並根據結果選擇適宜的壓片基質。
3.1.4 核磁共振(NMR) 本項測試可獲得藥物組成的某些元素在分子中的類型、數目、相互連接方式、周圍化學環境、甚至空間排列等信息,進而推測出化合物相應官能團的連接狀況及其初步的結構。常用的有氫核磁共振譜(1H-NMR)和碳核磁共振譜(13C-NMR)等。
核磁共振測試的重要參數有化學位移(δ )、偶合常數(J 值)、峯形、積分面積等。
溶劑峯或部分溶劑中的溶劑化水峯可能會對藥物結構中部分信號有干擾,因此測試時應選擇適宜的溶劑和方法,以使藥物所有信號得到充分顯示。
3.1.4.1 氫核磁共振譜(1H-NMR) 該項測試可提供供試品結構中氫原子的數目、周圍化學環境、相互間關係、空間排列等信息。此外,屬於 1H-NMR測試的 NOE(Nuclear Overhauser effect)或 NOESY 試驗,還可給出某些官能團在分子中位置、優勢構象及構型。
對含有活潑氫的藥物必需進行氘代實驗,以提供活潑氫的存在以及位置的信息。
3.1.4.2 碳核磁共振譜(13C-NMR) 該項測試可提供供試品結構中不同碳原子的類型以及所處的不同化學環境信息。
DEPT(Distortionless enhancement by polarization transfer)譜可進一步明確區分碳原子的類型,對於結構複雜的藥物,DEPT 譜對結構解析可給予更加有力的支持。
3.1.4.3 二維核磁共振譜 常用的二維核磁共振測試包括 H-Hcosy( H-H Correlated spectroscopy )、 HMBC ( 1H-detected multiple-bond heteronuclear multiple-quantum coherence )、 HMQC ( 1H-detected heteronuclear multiple-quantum coherence)等,對於結構複雜或用一般NMR 方法難以進行結構確證的化合物,進行二維譜測試可更有效地確證藥物的結構。
3.1.4.4 其它核磁共振譜 分子式中含 F、P 等元素的藥物,進行相應的 F、P 譜測試,除可提供相應元素的種類、在分子中所處的化學環境等信息外,對藥物元素組成測試亦有佐證作用。
3.1.5 質譜(MS) 用於原子量和分子量的測定、同位素的分析、定性或定量的分析,重要參數有分子離子峯、碎片峯、丰度等。
分子離子峯是確證藥物分子式的有力證據,應根據藥物自身結構特性選擇適宜的離子源和強度,同時儘可能地獲得分子離子峯和較多的、可反21 映出藥物結構特徵的碎片峯。
對含有同位素元素(如 Cl、Br 等)的藥物,利用分子離子峯及其相關峯丰度間的關係,可以判斷藥物中部分組成元素的種類、數量。
高分辨質譜是通過精確測定分子量確定藥物分子式,但它不能反映藥物的純度和結晶水、結晶溶劑、殘留溶劑等情況。 隨着科學的發展,在藥物研究中也採用了 GC-MS、MS-MS、LC-MS 等方法,研發者應根據藥物的組成和結構特徵選擇適宜的方法。
3.1.6 粉末 X-衍射(XRPD) 可用於固態單一化合物的鑑別與晶型確定,晶態與非晶態物質的判斷,多種化合物組成的多相(組分)體系中的組分(物相)分析(定性或定量),原料藥(晶型)的穩定性研究等。
手性藥物的結構(或通過生成其衍生物)確證應在上述一般研究的基礎上,對其絕對構型進行確證。常用方法有單晶 X-衍射(XRSD)、核磁共振譜(NMR)、圓二色譜(CD)、旋光光譜(ORD)以及前述的 NOESY 或NOE 譜(主要適用於具有剛性結構的藥物)等。其中單晶 X-衍射(XRSD)爲直接方法,後三種爲間接方法。
3.2.1 單晶 X-衍射(XRSD) 可獲得有關藥物晶型的相關信息、藥物的相對或絕對構型以及與藥物以結晶形式存在的水/溶劑及含量等一系列信息。
手性藥物絕對構型的測試,建議採用單晶 X 射線四園衍射儀,CuKα靶,衍射實驗的 θ 角範圍不低於 57°。
普通的單晶 X-衍射不能區分對映體,僅能推導出在空間的相對位置和藥物的相對構型。
3.2.2 圓二色譜(CD ) 該項測試通過測定光學活性物質(藥物)在圓偏振光下的 Cotton 效應,根據 Cotton 效應的符號獲得藥物結構中髮色團周圍環境的立體化學信息,並與一個絕對構型已知的與待測藥物結構相似藥物的 Cotton 效應相比較,即可能推導出待測物的絕對構型。
此外對於一般具有剛性結構的環體系的羰基藥物,通過比較其 Cotton效應的符號並結合經驗規律“八區律”,亦可能預測某些羰基藥物的絕對構型。
3.2.3 旋光光譜 通過比較相關藥物的旋光性,可得到手性藥物的相對構型信息。如能得知藥物旋光的可測範圍,則在一系列反應後,藥物絕對構型可從用於製備該藥物的底物構型推導得到。 在採用該方法測定藥物絕對構型時,要在相同的溶劑中以相同的濃度和溫度測定旋光,以保證比較的可靠性。
3.2.4 NOESY 或 NOE 譜 通過對具有剛性結構(或優勢構象)藥物官能團上質子的選擇性照射,致使與其相關質子峯強度的增減和相互間偶合作用的消失,從而推測出鄰近官能團的空間構象,進而可獲得藥物構型的信息。
3.3 藥物晶型的研究
在藥物研發過程中,多晶型現象是普遍存在的,其中有部分藥物因晶型不同具有不同的生物利用度和/或生物活性,特別是水溶性差的口服固體藥物。
對於新化學實體的藥物,應對其在不同結晶條件下(溶劑、溫度、結晶速率等)的晶型進行研究;通過不同晶型對藥物活性和毒性等影響的研究可爲其臨牀應用晶型的選擇提供依據。
對於仿製已上市的藥物,應進行自制藥物的晶型與已上市藥物晶型比較的研究,以保證自制品晶型的正確性。 進行連續多批樣品晶型一致性的研究,是判斷藥物製備工藝是否穩定的依據之一。
藥物晶型測定方法通常有粉末 X-衍射、紅外光譜、熱分析、熔點、光學顯微鏡法等。
3.3.1 粉末 X-衍射(XRPD ) 該項測試是判斷化合物(藥物)晶型的首選方法。
3.3.2 紅外光譜(IR) 結構相同但晶型不同的藥物其紅外光譜在某些區域可能存在一定的差異,因此比較藥物的 IR 可以用於區分藥物的晶型,但應注意在研磨、壓片時可能會發生藥物晶型的改變。
3.3.3 熔點(Melt point,mp) 結構相同但不同晶型的藥物其熔點可能存在一定的差異,熔點也可以用於晶型研究。
3.3.4 熱分析 用於藥物的物理常數、熔點和沸點的確定,並作爲鑑別和純度檢查的方法。晶型不同的藥物其熱分析圖譜有一定的差異,常用的方法有差示掃描量熱法(DSC)和差熱分析法(DTA)等。
對於含有結晶水或結晶溶劑的藥物,應對藥物中的水分/溶劑進行分析。
常用分析方法爲熱重、差熱分析、乾燥失重、水分測定、核磁共振以及單晶 X-衍射(XRSD)。
3.4.1 熱重 可獲得藥物的吸附水/溶劑、結晶水/溶劑及初步的分解溫度等信息。結合差熱分析的結果,還可判斷測試藥物在熔融時分解情況。
3.4.2 差熱分析 該項測試可推測出測試藥物的吸附水/溶劑、結晶水/溶劑以及熔點、有無多晶型存在和熱焓值等信息。
3.4.3 乾燥失重 該方法可以獲得藥物中的結晶水或溶劑、吸附水或溶劑的含量。
3.4.4 水分測定 可以獲得樣品中總含水量的信息(結晶水或吸附水)。
3.4.5 單晶 X-衍射(XRSD) 單晶 X-衍射在提供藥物元素組成、分子量及結構的同時,還可提供藥物中以結晶形式存在的水或溶劑的信息,包括結晶水或溶劑的種類、數量、存在方式等。
3.4.6 其他方法 如通過核磁共振測試,有可能獲得藥物中含有的部分結晶溶劑的信息。
以上分析方法均有各自的優、缺點,在藥物的結構確證研究中應根據藥物的結構特徵,選擇適宜的方法,同時也可利用不同方法所得結果進行相互補充、佐證,以確定存在藥物中水或溶劑的種類、數量和形式。
結構中含有金屬離子以及 F、P 等元素的藥物,可進行相應金屬原子吸收以及 F、P 等元素的測定。
3.5.1 原子發射光譜法和原子吸收分光光度法( Atomic emission spectrophotometry、Atomic absorption spectrophotometry,AES 、AAS)可用於含有多種金屬離子的藥物中無機微量元素的含量分析。 AES 常用於金屬元素的定性研究,AAS 可用於金屬元素定量研究。
3.5.2 絡合金屬離子存在方式的檢測 對於分子中含有順磁性金屬離子的藥物,常用的核磁共振(NMR)方法不能得到金屬離子在藥物中存在方式的確切信息,可採用單晶 X-衍射等方法進行檢測。
在結構確證研究中,參考文獻和結構確證用對照品對結構確證具有重要的佐證意義,但不是藥物結構確證研究的必要條件。
4.1 參考文獻對結構確證的意義和要求
參考文獻對藥物的結構確證具有重要的佐證作用,所用的參考文獻應引自國內外權威雜誌或專利,但應注意的是不同的測試條件所得到的測試結果亦可能有所差異。藥物不同研發階段的參考文獻對藥物結構確證所起到的佐證作用可能不同。
結構確證用對照品的結構信息對藥物的結構確證亦具有重要的佐證作用,不同來源的結構確證用對照品對藥物結構確證的佐證程度不同。對於從製劑中提取、精製所得的結構確證用對照品,如未能驗證在提取過程中晶型是否變化,此結構確證用對照品不能作爲晶型測定和與晶型有關的其它圖譜(如 IR、粉末 X—射線衍射)以及理化性質(如熔點、差熱分析、熱重分析)檢測的對照依據。
結構確證用對照品和測試樣品應在同一儀器上採用相同的測試條件進行測試,其純度應不低於精製品純度,以保證結構確證用對照品對藥物結構確證的支持。
5、綜合解析
以上每一種方法僅對藥物的結構研究提供相對分散的部分信息,需要通過綜合解析對這些信息進行綜合並全面分析,才能得到目的物完整的結構情況,綜合解析不應是對各項試驗結果的羅列。
對於新化學實體的藥物,由於沒有相關的文獻和對照品,單一的信息往往不能證明藥物的結構,需要對各種方法所得結果進行綜合分析,才能準確的解析藥物結構,包括絕對構型以及晶型、結晶水或結晶溶劑的情況。對於已有文獻報道的藥物,結構確證工作可相對簡單,特別是文獻數據或對照品的數據對結構確證具有一定的參考價值。
綜合解析應遵循簡明扼要、有機、合理、深入的原則。簡明扼要即是以簡潔的語言給出不同方法對藥物結構確證的結果,應避免過多的基本理論解說和繁雜的推導;有機是對不同方法所得的同一藥物結構不同方面信息的綜合歸納,以求獲得藥物較完整的結構信息;合理即是對數據進行合理的歸屬、解析,不牽強附會;深入即是在現有解析結果的基礎上,根據相互間的關係,獲得與藥物結構有關的更深層次的信息,以求得對藥物結構的完整認識。
6.1 藥物的名稱
藥物的名稱是藥物結構的正確反映,提供正確的、符合要求的藥物名稱有利於對藥物結構的認識和信息交流,藥物研發者在藥物研發的起始階段必須對所研製藥物的名稱有正確的認識,這裏所述的藥物的名稱包括化學名稱和通用名稱。
6.1.1 藥物的化學名稱 藥物的化學名稱分爲英文名稱和中文名稱,其名稱的制訂建議遵循 IUPAC 規則。
6.1.2 藥物的通用名稱 藥物的英文通用名稱應儘可能地參照國際非專利藥名(INN)確定;對 INN 未報道的藥品,可採用其他合適的英文名稱,但對結構與已報道或已上市的藥品結構屬同類藥物,通用名稱(後綴)應基本一致。 中文通用名稱應與國家制訂並頒佈的名稱一致。
6.2.1 分子式 分子式可提供藥物元素組成信息,爲制訂適宜的元素分析方法提供依據,如藥物分子中含有結晶水或結晶溶劑,應以適宜的形式註明。
6.2.2 分子量 分子量既可對分子式進行驗證並作爲元素分析的基礎,也有助於質譜的解析,其精確程度應根據所測試質譜的類型而定。
6.3 結構式 結構式是藥物結構的具體存在形式,提供正確的結構式是結構確證研究的目的,有助於深入理解藥物的結構、性質和測試方案的制訂。
對於存在異構體、含有結晶水或溶劑、手性中心、絡合離子、酸根和鹼基的藥物,應在結構式中注明其異構的形式、手性中心的絕對構型、絡合位置/方式、酸根/鹼基和結晶水或溶劑的位置。
6.4 物理常數
物理常數是反映藥物物理性質的重要數據。對藥物物理常數的研究有助於瞭解藥物的物理性質,併爲藥物的質量控制研究服務。
藥物的物理常數一般包括熔點、沸點、沸程、凝點、折光率、粘度、相對密度、溶解度、比旋度、紫外吸收係數等。
對於已有文獻報道的藥物,將其物理常數的測試結果與文獻報道值比較,對藥物的結構確證亦是有力的支持。
3.4 (四)名詞解釋
結構確證用對照品:是指申報藥物爲已批准上市的藥物時,從有合法生產資格非申報單位得到的符合標準的樣品,包括源自藥檢所的樣品、試劑公司的化合物等。
精製品:是按申報資料中所用生產工藝所製得結構單一製品或在此基礎上經進一步純化後所得樣品。
富集異構體:是指在一對對映體混合物或非對映體混合物中,某一對映體或非對映體過量的化合物。
3.5 (五)參考文獻
1、FDA :Guidance for Industry Drug Substance,Chemistry Manufacturing,and Controls Information,2004.
2、EMEA :Note For Guidance On Chemistry of The New Active Substance, 2003.
3、林國強 等. 手性合成——不對稱合成及其應用。 北京科學出版社,2000 年版。
4、馬廣慈,唐任寰,鄭斯成等. 藥物分析方法與應用。 北京科學出版社, 2000 第一版。
5、鄭筱萸. 《化學藥品和治療用生物製品研究指導原則(試行)》。中國醫藥科技出版社, 2002 第一版。
4 三、著者
《化學藥物原料藥製備和結構確證研究的技術指導原則》課題研究組